RESUMEN
La poliquistosis renal autosómica dominante (PQRAD) es la causa más frecuente de nefropatía genética y representa entre el 6 y el 10% de los pacientes en terapia de reemplazo renal (TRR).
Muy pocos ensayos prospectivos, aleatorizados o estudios clínicos abordan el diagnóstico y el tratamiento de este trastorno relativamente frecuente. No hay guías clínicas disponibles hasta la fecha. Este es un documento de consenso revisada de la versión anterior del 2014, que presenta las recomendaciones del Grupo de Trabajo Español de Enfermedades Renales Hereditarias, acordadas tras la búsqueda bibliográfica y discusiones. Los niveles de evidencia en su mayoría son C y D según el Centro de Medicina Basada en Evidencia (Universidad de Oxford). Las recomendaciones se relacionan, entre otros temas, con el uso de diagnóstico por imágenes y genético, el manejo de la hipertensión, el dolor, las infecciones y el sangrado quístico, la afectación extrarrenal, incluida la enfermedad poliquística hepática y los aneurismas craneales, el manejo de la enfermedad renal crónica y el TRR, así como el seguimiento de niños con PQRAD. Se proporcionan recomendaciones sobre terapias específicas para la PQRAD, así como la recomendación para evaluar la rápida progresión.
Introducción
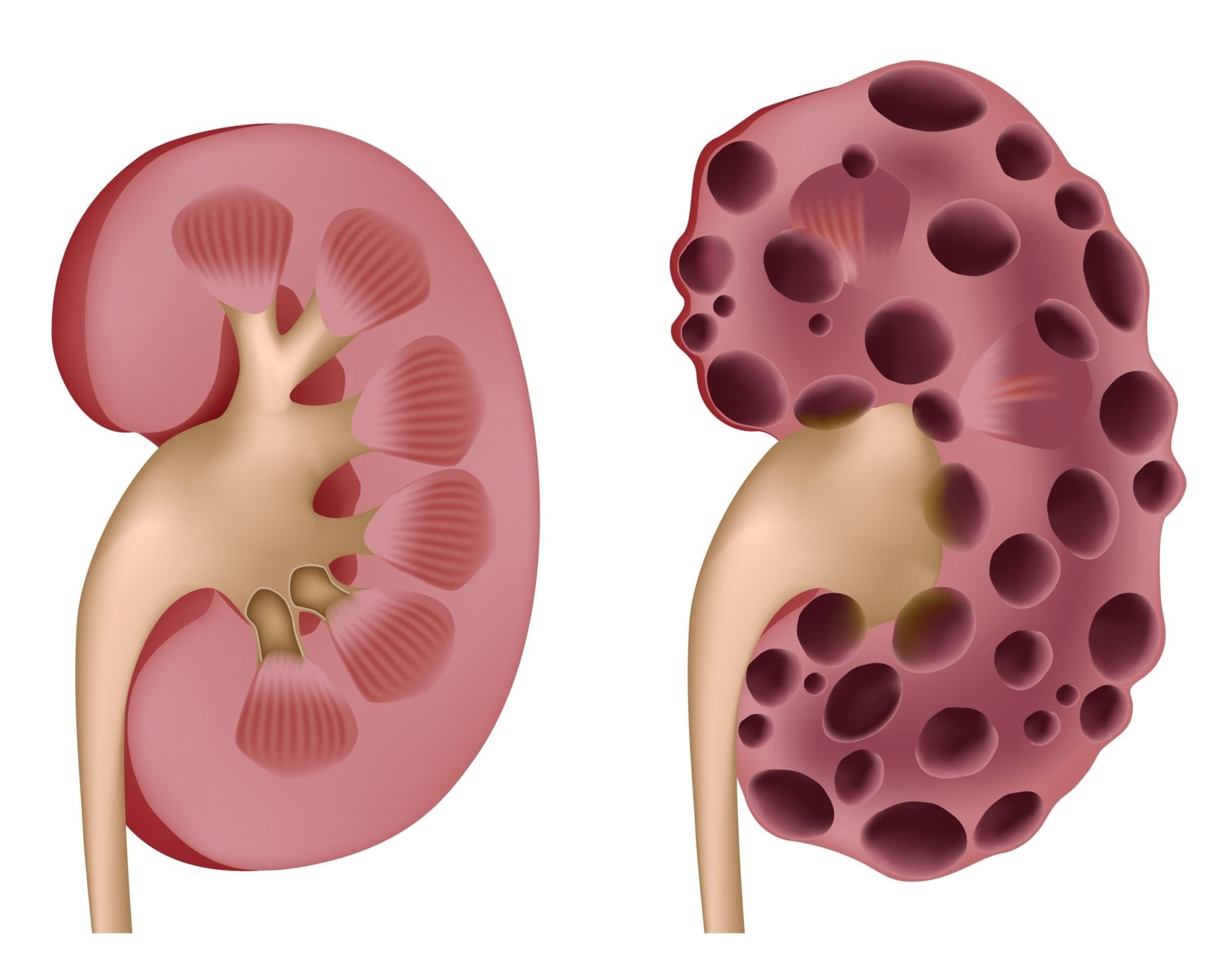
La poliquistosis renal autosómica dominante (PQRAD) es la enfermedad renal hereditaria más frecuente. Su prevalencia estimada está muy discutida y oscila entre de 1 cada 500 personas y una de cada 20001-4.
Los pacientes con PQRAD constituyen entre un 6 y un 10%, aproximadamente, de la población en diálisis o trasplante renal, siendo, por lo tanto, una enfermedad con un gran impacto social5,6. Se caracteriza por la progresiva aparición de quistes renales que suelen conducir a la ERC terminal (ERCT), generalmente en la edad adulta. Así mismo se asocia a manifestaciones sistémicas, tales como hipertensión arterial (HTA), aneurismas intracraneales (AIC), poliquistosis hepática, anomalías valvulares y quistes en otros órganos. Durante las últimas 3 décadas se han producido grandes avances en el conocimiento de la enfermedad. A mitad de los años 90 se identificaron los genes causantes de la PQRAD: PKD1 y PKD7,8. Se han identificado otros genes causantes de nefropatía quística autosómica dominante que clínicamente difieren de la PQRAD (GANAB, DNAJB11)9,10. PKD1 y PKD2 codifican las poliquistinas 1 y 2, que se localizan en el cilio primario11. Las moléculas sobreexpresadas o deficitarias en las células poliquísticas se convirtieron en potenciales dianas terapéuticas, de manera que actualmente hay varios fármacos en estudio como tratamiento de la enfermedad. En 2014 se aprobó en Japón y en 2015 en Canadá y Europa la utilización de tolvaptán para el tratamiento de los pacientes mayores de 18 años con PQRAD en estadios 1-3 al inicio del tratamiento y signos de progresión rápida de la enfermedad. En junio del 2018 la Agencia Europea del Medicamento (EMA) amplió las indicaciones de tolvaptán a pacientes con PQRAD y estadios 1-4 al inicio del tratamiento y evidencia de rápida progresión (https://www.ema.europa.eu/en/documents/procedural-steps-after/jinarc-epar-procedural-steps-taken-scientific-information-after-authorisation_en.pdf) y el mismo año fue aprobado en Estados Unidos para adultos rápidos progresadores sin definir estadio de ERC al inicio del tratamiento12,13. Existen escasos ensayos controlados aleatorios prospectivos, así como pocos estudios clínicos que incorporen un diseño experimental para el diagnóstico y el manejo de la PQRAD. Dichos estudios son difíciles de realizar debido al número relativamente bajo de pacientes en cada centro, así como a la heterogeneidad de la presentación clínica de la enfermedad. Las recomendaciones de consenso de las presentes guías clínicas se basan en una búsqueda en la literatura y, en gran medida, en la experiencia y las opiniones de los autores. Se han utilizado la Cochrane Library, MEDLINE y la base de datos de Revisiones Sistemáticas (hasta el 1 de julio del 2019). Se hicieron búsquedas utilizando los términos de búsqueda «PQRAD» o «riñón poliquístico» en combinación con los términos «diagnóstico» o «imágenes» o «gen» o «HTA» o «ERC» o «enfermedad renal crónica» o «insuficiencia renal crónica» o «ERCT» o «enfermedad renal crónica terminal» o «diálisis» o «trasplante» o «infección» o «dolor» o «hígado» o «aneurisma o «cáncer» o «embarazo» o «niños» o «tolvaptán». En gran medida, seleccionamos las publicaciones de los últimos 10 años, pero no se excluyeron las publicaciones más antiguas relevantes. Se han tenido especialmente en cuenta las recientes KDIGO (Kidney Disease Improved Global Outcomes) en PQRAD14. También se revisaron las listas de referencias de los artículos identificados y seleccionaron aquellos que se juzgó pertinente. Los artículos de revisión se citan con el fin de ofrecer a los lectores más detalles de los que se exponen en estas guías clínicas. Los autores han pretendido que esta guía sea concisa y muy práctica y que uniformice, en la medida de lo posible, la asistencia a los pacientes con PQRAD.
Los autores son miembros del Grupo de Trabajo de Enfermedades Renales Hereditarias de la Sociedad Española de Nefrología. Firman las guías en orden alfabético, menos el último autor. Los autores llegaron a un consenso sobre las recomendaciones y consideraron que los beneficios superaban los riesgos potenciales. El documento ha sido sometido a revisión pública por parte de los miembros de la Sociedad Española de Nefrología. Los niveles de evidencia son mayoritariamente bajos: niveles C y D de acuerdo con el Centro para la Medicina Basada en la Evidencia (Universidad de Oxford) (http://www.cebm.net/?o=1025). En estas guías se abordan los siguientes aspectos de la enfermedad: diagnóstico, HTA, dolor, evaluación de la progresión de la enfermedad renal, enfermedad renal terminal, poliquistosis hepática, AIC, otras características extrarrenales, tratamiento específico de la enfermedad y PQRAD en los niños. La versión inicial de estas guías fue publicada en el año 2014 y actualizada en 201715.
Elisabet Arsa, Carmen Bernisb, Gloria Fragac, Mónica Furlanod, Víctor Martíneze, Judith Martinsf, Alberto Ortizg, Maria Vanessa Pérez-Gómezg, José Carlos Rodríguez-Pérezh, Laia Sansi, Roser Torrad,
, en nombre del grupo de trabajo de Enfermedades Renales Hereditarias de la Sociedad Española de Nefrología
a Laboratorio de Biología Molecular, Fundació Puigvert, Instituto de Investigaciones Biomédicas Sant Pau (IIB-Sant Pau), Universitat Autònoma de Barcelona, REDinREN, Instituto de Investigación Carlos III, Barcelona, España
b Servicio de Nefrología, Hospital de la Princesa, REDinREN, Instituto de Investigación Carlos III, Madrid, España
c Sección de Nefrología Pediátrica, Hospital de la Santa Creu i Sant Pau, Universidad Autónoma de Barcelona, Barcelona, España
d Enfermedades Renales Hereditarias, Servicio de Nefrología, Fundació Puigvert, Instituto de Investigaciones Biomédicas Sant Pau (IIB-Sant Pau), Universidad Autónoma de Barcelona (Departamento de Medicina), REDinREN, Barcelona, España
e Servicio de Nefrología, Hospital Virgen de la Arrixaca, Murcia, España
f Servicio de Nefrología, Hospital Universitario de Getafe, Universidad Europea de Madrid, Getafe, Madrid, España
g Servicio de Nefrología, IIS-Fundación Jiménez Díaz, Universidad Autónoma de Madrid, IRSIN, REDinREN, Madrid, España
h Servicio de Nefrología, Hospital Universitario de Gran Canaria Dr. Negrín, Universidad de Las Palmas de Gran Canaria, Las Palmas de Gran Canaria, Las Palmas, España
i Servicio de Nefrología, REDinREN, Instituto de Investigación Carlos III, Hospital del Mar, Barcelona, España
Para descargar la investigación completa haga clik a continuación: